集产品研发、生产、销售于一体
多年产品研发经验,自主知识产权
13671881166
400-6885-928

集产品研发、生产、销售于一体
多年产品研发经验,自主知识产权
13671881166
400-6885-928

众所周知,一氧化碳(CO)是一种没有颜色、没有气味,而在高浓度时具有毒性的气体。它是碳和含碳化合物不完全、不充分燃烧的副产品,是内燃机排出的主要污染物之一,是各种环境和健康问题的根源 [1]。随着世界各国交通运输业和工矿企业的不断发展,煤炭和石油燃料的消耗量不断增加,一氧化碳排放量也在不断增加 [2] [3]。此外,人类过量吸入一氧化碳气体会引起缺氧,甚至会造成死亡 [4] [5]。因此,如何降低大气中一氧化碳的浓度引起了广大科研工作者的广泛关注。在众多方法中,催化氧化CO被认为是最有效的方法之一 [6] [7] [8]。
虽然CO氧化反应是一个简单的化学反应,但它可以作为模型反应来评价非均相催化剂的性能,研究催化剂的结构和表面性质、反应机理以及金属–载体相互作用 [9] [10]。CO氧化反应不仅在基础科学中非常重要,而且在机动车尾气处理 [10] [11]、质子交换膜燃料电池(PEMFC) [10] [12]、空气净化 [9] [10] 等领域的应用也十分广泛。在现有的文献报道当中,用于CO氧化反应的催化剂主要有金属氧化物催化剂、贵金属催化剂和单原子催化剂 [13] [14] [15] [16]。
金属氧化物催化剂通常为复合氧化物(complex oxides),即多组分的氧化物,例如V2O5-MoO3 [17],TiO2-V2O5-P2O5 [18],V2O5-MoO3-A12O3 [19] 等等。在1997年,Luo等人 [20] 制备了不同CuO负载量的CuO/CeO2催化剂,并将其用于反应物CO和O2为化学计量比的CO氧化反应中。通过X射线衍射(XRD)、氢气程序升温还原(H2-TPR)和一氧化碳程序升温脱附(CO-TPD)等对催化剂进行了表征。结果表明,相比于Cu-Ce-O复合催化剂,浸渍法制备的CuO/CeO2催化剂具有更高的CO氧化活性,催化活性也十分稳定。CuO/CeO2催化剂的H2-TPR研究表明,CeO2可以促进CuO物种的还原,在H2-TPR谱图中观察到两个还原峰,表明存在两种不同的CuO物种,CuO/CeO2的低温TPR峰归属于能吸附CO的高分散CuO物种,而高温峰归属于不能吸附CO的块状CuO物种。因此,提出了用浸渍法制备的CuO/CeO2催化剂,在低温下可以被还原、能够吸附CO的高分散CuO物种对低温区CO氧化活性贡献较大,在高温下可以被还原、不能吸附CO的块状CuO对CO氧化活性的贡献很小,高度分散在CeO2表面上的CuO有利于低温区CO氧化。
然而,对同一种复合型金属氧化物催化剂来说,不同的煅烧温度、不同的负载量往往也会对催化剂的结构产生影响,从而进一步对催化剂催化CO氧化反应的催化作用产生不同的效果。Huang等人 [21] 研究了沉积–沉淀(DP)法制备的CuO/TiO2催化剂,并将其用于CO氧化反应研究。通过热重–差热分析(TG-DTA)、X射线衍射(XRD)和X射线光电子能谱(XPS)测试对比研究了CuO/TiO2催化剂的结构特征。TiO2颗粒充当载体,CuO充当活性组分。结果表明,CuO/TiO2催化剂的催化性能在很大程度上取决于TiO2载体的焙烧温度、CuO负载量和CuO/TiO2催化剂的焙烧温度。CuO/TiO2催化剂中载体TiO2的煅烧温度为400℃时的CO氧化活性比在500℃和700℃下煅烧的催化活性更好。在CuO负载量为2%~12%的所有催化剂中,CuO负载量为8 wt%的催化剂的催化活性最好。CuO/TiO2催化剂的最佳焙烧温度为300℃。因此可以得出CuO/TiO2催化剂的催化活性取决于以下因素:1) 二氧化钛载体的煅烧温度。催化活性强烈依赖于TiO2载体的煅烧温度,也就是说,催化活性受到TiO2载体的晶相和粒度的影响。在400℃煅烧的TiO2载体上负载CuO在CO氧化反应中表现出最好的催化活性。2) 氧化铜的负载量。CuO负载量为8 wt%的CuO/TiO2催化剂的催化活性远高于其他CuO负载量的催化剂,这可能是因为8 wt%的CuO负载量最接近在TiO2上单层分散CuO的阈值(7.3 wt%)。3) CuO/TiO2催化剂的煅烧温度。在200℃~300℃的煅烧温度区间内,CuO/TiO2催化剂的CO氧化活性差别可以归因于CuO完全结晶,而随着煅烧温度从300℃升高到500℃,催化活性降低可能是由于CuO晶粒尺寸的增加。
对于单组分金属氧化物催化剂而言,Jansson等人 [22] 用流动反应器实验、原位光谱和结构方法研究了室温下CO在Co3O4上的氧化。Co3O4在室温稳态CO氧化过程中缓慢失活。失活速率随着CO或CO2气体浓度的增加而增加,但失活速率随着O2浓度的增加或温度的升高而降低。催化剂在10% O2/Ar中的再生效果比单独在Ar中的再生效果好。根据程序升温氧化(TPO)实验推断,失活催化剂的表面存在碳酸盐和表面羰基。然而,这些物种都与催化剂的失活无关。原位红外证明了表面碳酸盐、羰基和氧物种的存在。通过原位X射线衍射(in-situ XRD)、原位X射线吸收近边结构(in-situ XANES)、X射线光电子能谱(XPS)和流动反应器实验研究了催化剂结构和氧化态的变化。Jansson等人 [22] 认为活性的降低是由于氧化钴的表面重建,使得钴离子对CO吸附不起作用。
水汽的存在往往会使CO氧化反应催化剂产生较为明显的失活,从而影响催化活性。申文杰研究员等人 [23] 利用纳米催化材料的形貌效应,使金属氧化物能够较多地暴露高活性晶面,从而表现出很好的CO氧化性能。通过对制备条件的精确调控,成功地制备出了结构规整的Co3O4纳米棒,其中活性(110)晶面占纳米棒表面的40%以上。由于(110)晶面含有较多的Co3+活性位点,即使在−77℃、水汽存在的条件下仍然可以实现CO的完全转化,其反应速率是普通Co3O4纳米粒子的10倍以上。这类Co3O4纳米棒在接近汽车发动机冷启动的条件下表现出非常好的CO氧化性能和结构稳定性。这种通过形貌控制优先暴露活性晶面的方法还可以适用于其他金属氧化物体系,这一研究成果对纳米催化的基础研究和开发新一代高活性的氧化催化剂具有重要的借鉴价值。
贵金属催化剂主要是以铂族金属为主,主要包括铂(Pt)、钯(Pd)、钌(Ru)、铑(Rh)、铱(Ir)、锇(Os)等为催化活性组分的负载型非均相催化剂和铂族金属无机化合物或有机金属配合物组成的各类均相催化剂。铂族金属由于其d电子轨道都未填满,表面易吸附反应物,且吸附强度适中,有利于形成中间“活性化合物”,具有较高的催化活性,同时还具有耐高温、抗氧化、耐腐蚀等综合优良特性,因此是催化领域重要的催化剂材料之一。贵金属催化剂在石油、化工、医药、农药、食品、环保、能源、电子等领域中占有极其重要的地位,同时贵金属催化剂在汽车尾气净化、有机物催化燃烧、CO、NO氧化等环保领域的应用也十分广泛。
在1996年,Mergler等人 [24] 介绍了以MnOx和CoOx为载体的Pt基催化剂在CO氧化反应中的研究。将Pt/SiO2、Pt/MnOx/SiO2和Pt/CoOx/SiO2催化剂的催化活性与工业催化剂Pt/Al2O3、Pt/Rh/Al2O3和Pt/CeOx/Al2O3的催化活性进行比较。结果表明一系列催化剂的CO氧化活性顺序为:Pt/CoOx/SiO2 > Pt/MnOx/SiO2,Pt/CeOx/Al2O3 > Pt/Al2O3,Pt/Rh/Al2O3,Pt/SiO2。同时Pt/CoOx/SiO2催化剂在室温下就可以将CO氧化。此外Mergler等人 [24] 还讨论了Pt/CoOx/SiO2催化剂的CO氧化活性较高的原因,可能是由于CoOx上的O空位可以作为O2的解离中心,同时,部分还原的金属氧化物能够提高Pt/CoOx/SiO2,Pt/MnOx/SiO2和Pt/CeOx/Al2O3催化剂的CO氧化催化活性。
在贵金属催化剂中不止是贵金属会与载体之间产生一定的相互作用,贵金属与其他金属之间也会互相影响。在2010年,Xu等人 [25] 研究了以二氧化硅为载体的Pd-Au纳米合金催化剂,用于催化低温CO氧化反应。除纯金属外,这些材料总是表现出两相分离,形成富含钯或富含金的材料。研究结果表明,Pd4Au1催化剂的CO氧化反应性能是最好的,这种催化剂是Pd90Au10 (72.5 at.%)和Pd31Au69 (27.5 at.%)的混合物,在300 K下具有优异的催化活性,并且比纯金催化剂更稳定。对于以Pd作为主体材料的催化剂(Pd:Au = 16:1; 8:1; 4:1),富钯合金组分通常以空心球或环形形态存在,而富金晶体通常为多孪晶晶体。X射线衍射(XRD)表明,在所用的合成条件下,金在钯晶体中的溶解度极限为12 at.%,而Pd更易溶于Au (~31 at.%),这与X射线光电子能谱(XPS)相一致,揭示了富Pd合金中的Au主要集中在合金表面,同时合金化电子修饰Pd和Au元素可以改变Pd3d和Au4f核心能级并促进Pd和Au之间的电荷转移,从而有利于CO的吸附。
由于贵金属资源稀少、价格昂贵,人们也在不断研究如何可控的合成贵金属催化剂以减少贵金属的用量。Qadir等人 [26] 采用多元醇法合成了尺寸均匀的Ru纳米颗粒,尺寸分别为2.8 nm和6 nm。在钌纳米颗粒上进行催化氧化、还原和共氧化,并使用常压X射线光电子能谱(APXPS)测量和监测表面氧化状态。并且发现在CO氧化的过程中,较小的钌纳米颗粒在表面形成块状氧化钌,这是催化活性降低的原因。随着纳米颗粒尺寸的增加,RuO2的核壳结构变得稳定。表明钌纳米颗粒在CO氧化下催化活性提高可能与在较大的纳米颗粒上形成的活性超薄表面氧化物的高稳定性有关。Qadir等人 [26] 的研究显示了一种在现实条件下研究催化剂表面的独特方法,可以密切监控每个反应步骤中实时发生的变化。这种对钌纳米颗粒的原位观察有助于确定催化剂在使用过程中的活性状态,可以为实际应用进行合理的催化剂设计(图1)。
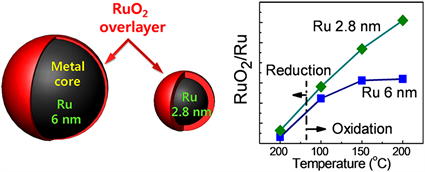
Figure 1. Schematic diagram of RuO2 core-shell structure and state at different temperatures [26]
图1. RuO2核壳结构示意图及不同温度下的状态
“单原子催化”的概念由中国科学院大连化学物理研究所的张涛院士、清华大学李隽教授及美国亚利桑那州立大学刘景月教授于2011年共同提出 [16]。单原子催化剂是一种特殊的负载型金属催化剂,专指载体上的所有金属组分都以单原子分散的形式存在,不存在同原子金属–金属键。在2014年后,单原子催化剂迅速成为催化领域的研究热点。
Qiao等人 [27] 开发了一种以Co3O4为载体的金单原子催化剂,其金含量非常低(0.05 wt%)。该催化剂表现出极高的CO氧化活性,并且在室温下就可以氧化CO,并产生约25 molco∙gAu−1∙h−1的比速率。将CO氧化的高活性归因于分布在Co3O4纳米晶上的孤立金原子,催化机理仍不是很明了。从图2中可以很清晰的看出单原子Au对CO氧化反应的影响,在金的担载量极低的情况下得到的高催化活性,证明了金单原子催化剂的高原子效率,并表明了金单原子催化剂的潜在应用前景。
之前的研究者们虽然通过实验可以证明单原子催化剂具有较高的CO氧化反应的催化活性,但对于单原子催化CO氧化反应的催化机理还知之甚少。Han等人 [28] 成功地开发了ZnO纳米线(ZnOnw)负载Rh、Au和Pt SAC催化剂,以研究它们的CO氧化性能。其中Rh1/ZnO-nwSAC显示出最高的CO氧化活性。DFT计算表明,该反应的反应机理可能是Mars-van Krevelen机理,表面氧空位处的O2离解是速率决定步骤。在所研究的三种贵金属中,Rh1/ZnO-nwSAC具有最低的限速步骤能垒,因此提供了最高的CO氧化活性。此外,Rh1/ZnO-nwSAC催化剂具有耐高温和抗烧结的性能使其成为实际应用的潜在候选材料。
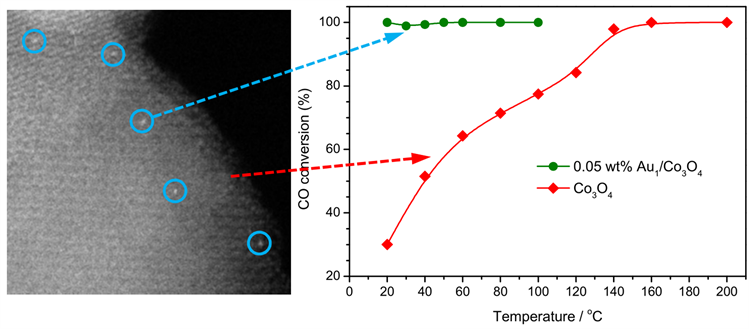
Figure 2. Comparison of catalytic activity between Au1/Co3O4and Co3O4 [27]
图2. Au1/Co3O4与Co3O4的催化活性对比 [27]
Muravev等人 [29] 比较了两种催化低温CO氧化反应的Pd/CeO2单原子催化剂,它们在反应条件下表现出截然不同的结构动力学。CeO2是通过水热法合成的,两种Pd/CeO2单原子催化剂分别是使用浸渍法和火焰喷雾热解法进行制备的。通过浸渍法制备的Pd/CeO2单原子催化剂表面分散的离子Pd在CO氧化过程中迅速还原并聚集成金属簇,如图2。火焰喷射热解法制备的催化剂上的表面Pd单原子在CO氧化条件下表现出高抗烧结性(图2)。结合原位近环境压力X射线光电子能谱(NAP-XPS)、原位漫反射傅立叶变换红外光谱(DRIFTS)和扩展X射线吸收精细结构(EXAFS)对瞬态和稳态CO氧化动力学分析结果表明,分散的表面Pd2+位点是两种催化剂可以在低温下催化CO氧化反应的原因,而还原的Pdδ+/0物种在高温下比较活跃。遵循Ce4+/Ce3+和Pd2+/Pdδ+/0氧化还原动力学,确定了Pd离子的掺杂活化了CeO2载体的晶格氧,并促进了Pd-O-Ce界面处的反向氧溢出。金属–载体界面处氧的流动性对单原子钯的稳定性和反应性有很大影响。这些发现为合理设计高活性和高稳定性的单原子催化剂开辟新的途径。
金属氧化物催化剂虽然会对CO氧化反应的活性有一定的提升,但金属的利用率不高,催化效率不是特别理想。贵金属催化剂很好的弥补了金属氧化物催化剂金属利用率不高的这一问题,而且对CO氧化反应的活性也有较大提升,但贵金属催化剂中贵金属价格比较昂贵,贵金属的原子利用率不高。单原子催化剂有效的提高了贵金属的原子利用率,并且能够在低温下有效催化CO氧化反应。总之,开发高效、经济、耐高温的CO催化氧化催化剂是该领域未来的发展趋势和研究热点。
浏览次数:149
浏览次数:388
浏览次数:362
浏览次数:453
浏览次数:488
